U.S. Food and Drug Administration06.02.17
The FDA has identified this as a Class I recall, the most serious type of recall. Use of these devices may cause serious injuries or death.
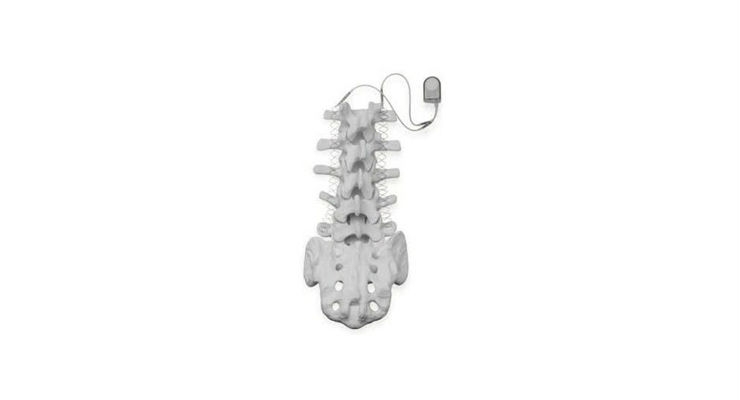
The Zimmer Biomet SpF PLUS-Mini and SpF XL IIb Implantable Spinal Fusion Stimulators are used during spinal fusion surgery to increase the possibility of permanently connecting two or more bones of the spine (backbone) together. The device is implanted into the patient's back and provides constant electrical stimulation to the surgical site.
Zimmer Biomet is recalling the SpF PLUS-Mini and SpF XL IIb Implantable Spinal Fusion Stimulators due to higher than allowed levels of potential harmful chemicals, which may be toxic to tissues and organs (cytotoxicity) and that were found during the company's routine monitoring procedure. A cytotoxicity test is a part of the biological evaluation of medical devices to ensure compatibility with the device and the human body. A positive cytotoxicity test (failed result) can indicate that a device contains potential harmful chemicals at amounts or levels that could be dangerous to the patient.
The use of affected product may cause serious adverse health consequences, including but not limited to chronic infections, long-term hospitalization due to additional surgical procedures, paralysis, and death.
Healthcare providers using the SpF PLUS-Mini and SpF XL IIb Implantable Spinal Fusion Stimulators, as well as all patients undergoing spinal fusion procedures involving the SpF PLUS-Mini and SpF XL IIb Implantable Spinal Fusion Stimulators may be affected.
On April 20, 2017, Zimmer Biomet sent an Urgent Medical Device Removal notification to all affected customers. The notice instructed customers to:
Customers with questions related to this recall are instructed to contact Zimmer Biomet at 1-800-447-3625 (Monday - Friday, 8 am - 5 pm Mountain Standard Time) or by email to corporatequality.postmarket@zimmerbiomet.com.
The Zimmer Biomet SpF PLUS-Mini and SpF XL IIb Implantable Spinal Fusion Stimulators are used during spinal fusion surgery to increase the possibility of permanently connecting two or more bones of the spine (backbone) together. The device is implanted into the patient's back and provides constant electrical stimulation to the surgical site.
Zimmer Biomet is recalling the SpF PLUS-Mini and SpF XL IIb Implantable Spinal Fusion Stimulators due to higher than allowed levels of potential harmful chemicals, which may be toxic to tissues and organs (cytotoxicity) and that were found during the company's routine monitoring procedure. A cytotoxicity test is a part of the biological evaluation of medical devices to ensure compatibility with the device and the human body. A positive cytotoxicity test (failed result) can indicate that a device contains potential harmful chemicals at amounts or levels that could be dangerous to the patient.
The use of affected product may cause serious adverse health consequences, including but not limited to chronic infections, long-term hospitalization due to additional surgical procedures, paralysis, and death.
Healthcare providers using the SpF PLUS-Mini and SpF XL IIb Implantable Spinal Fusion Stimulators, as well as all patients undergoing spinal fusion procedures involving the SpF PLUS-Mini and SpF XL IIb Implantable Spinal Fusion Stimulators may be affected.
On April 20, 2017, Zimmer Biomet sent an Urgent Medical Device Removal notification to all affected customers. The notice instructed customers to:
- Review notification and distribute the information to all appropriate personnel.
- Quarantine all affected products.
- The Zimmer Biomet sales representative will remove the affected product from the facility.
- If the product was purchased directly from the company, follow the instructions on the Certificate of Acknowledgment form provided by Zimmer Biomet to complete the return.
- Return the form to corporatequality.postmarket@zimmerbiomet.com or via fax to 574-372-4265.
- Retain a copy of the Acknowledgement Form with the field action records.
- Surgeons are reminded that normal clinical monitoring is recommended for 3-6 months post operatively for any patient with the affected devices implanted.
Customers with questions related to this recall are instructed to contact Zimmer Biomet at 1-800-447-3625 (Monday - Friday, 8 am - 5 pm Mountain Standard Time) or by email to corporatequality.postmarket@zimmerbiomet.com.